


Décodage des génomes du SARS-CoV-2 – Origine
La pandémie actuelle de COVID-19 est une menace mondiale pour les systèmes de santé publique et a durement touché l'économie mondiale. Le coupable de cette pandémie, le SRAS-CoV-2, n'est pas la grippe typique. Ce virus affecte à la fois les voies respiratoires supérieures et inférieures, interférant avec le processus vital de la vie, la respiration, et est donc mortel. Au 6 avril 2020, Worldometer avait signalé 1 337 166 cas et 74 176 décès dans le monde.
L'examen du SRAS-CoV-2 au niveau du génome permettra de mieux comprendre les origines de ce virus. Il aidera également les scientifiques à concevoir des outils de diagnostic pour détecter cet agent pathogène invisible et faciliter l'invention de thérapies pour minimiser les pertes de vie.
Comprendre le génome du SRAS-CoV-2
Un virus est un agent infectieux qui nécessite un hôte vivant pour prospérer et se répliquer. En outre, le SRAS-COV-2 est un virus à ARN simple brin avec un génome de près de 30 kb de bases nucléotidiques avec 12 cadres de lecture ouverts putatifs. Peu de temps après le début de l'épidémie en décembre 2019, des scientifiques chinois ont séquencé le génome du SRAS-CoV-2. Divers groupes scientifiques ont publié des séquences génomiques complètes du SRAS-CoV-2 au cours des dernières semaines. Celles-ci sont accessibles au public dans Genbank et dans la base de données sur les coronavirus.
Origine du virus SARS-CoV-2
Lors d'épidémies comme celle-ci, les théories du complot non scientifiques peuvent entraîner des préjugés inutiles contre les pays, les communautés et les cultures. Le SARS-CoV-2 ne fait pas exception, et la situation n'est qu'exacerbée par les plateformes de médias sociaux qui se multiplient aujourd'hui. Il nous incombe de voir cet ennemi invisible à travers une lentille scientifique rationnelle. Sur la base d'analyses du génome, le SRAS-CoV-2 est un virus qui a évolué naturellement et n'est pas une souche de laboratoire synthétique 1,2 . Les scientifiques ont séquencé les génomes complets de plus de 100 souches de SARS-CoV-2 collectées dans différentes régions du monde. Il s'avère que ces souches sont identiques à plus de 99,5% au niveau nucléotidique. Cela indique que les souches n'ont pas beaucoup muté dans différentes régions, apparemment car le virus a déjà un taux d'infection et une virulence élevés.
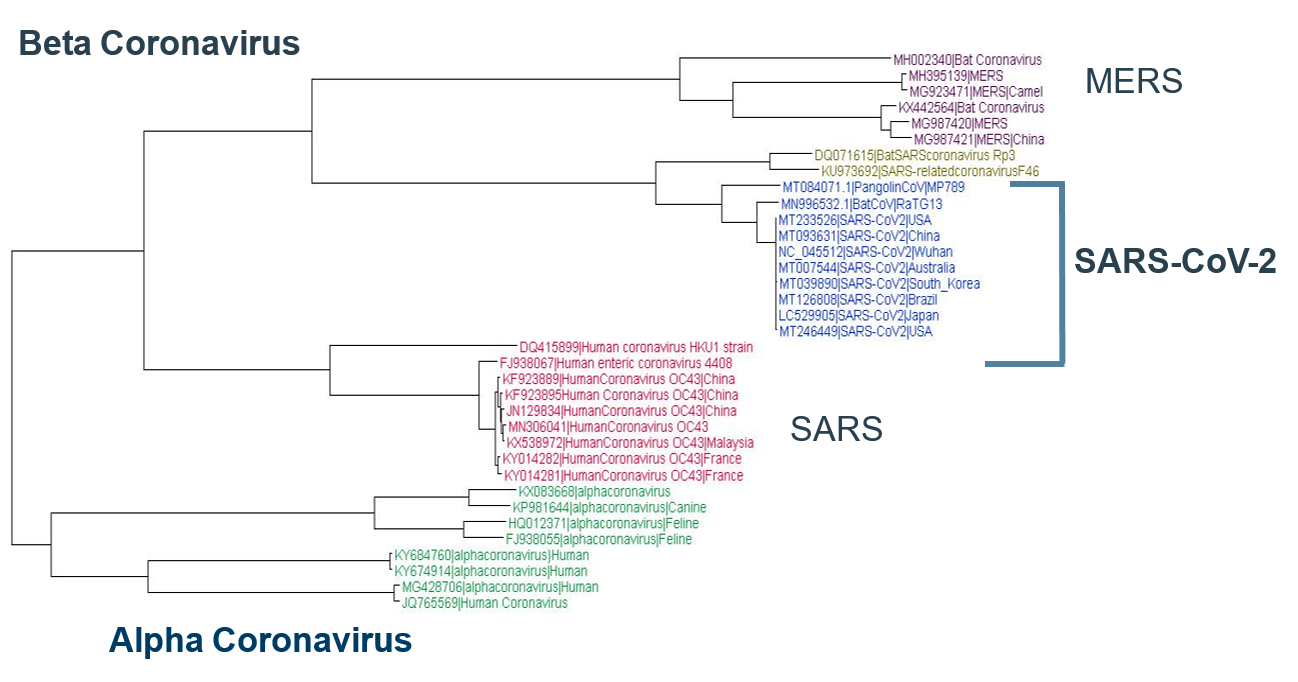
Dans un passé récent, deux autres coronavirus ont attiré l'attention du monde entier. Il s'agissait du SARS-CoV, Chine, 2002, et du MERS-CoV, Arabie saoudite, 2012. Il a été démontré que ces deux virus antérieurs provenaient de chauves-souris. Sur la base de ces connaissances historiques, les scientifiques ont séquencé le coronavirus des chauves-souris et ont montré que le Bat CoV (RaTG13 ) était identique à 96,2 % au SRAS-COV-2, confirmant ainsi l'origine zoonotique de ce dernier. 2 Le coronavirus utilise souvent un porteur intermédiaire avant d'infester l'homme. Fait intéressant, vers octobre 2019, des rapports faisant état de pangolins malais morts présentant des poumons et des symptômes de fibrose écumeuse pulmonaire au centre de sauvetage de la faune du Guangdong en Chine ont incité les scientifiques à isoler leur métagénome. En effet, les données du métagénome des pangolins morts contenaient le coronavirus ! 3
Fait intéressant, au niveau du génome entier, le SRAS-CoV-2 est presque identique à 91 % au Malayan Pangolin CoV, ce qui indique que les Pangolins pourraient être un hôte intermédiaire.
Que sont les pangolins ? Ce sont des mammifères fourmis très demandés en Asie pour une utilisation en médecine traditionnelle chinoise ainsi que pour leur viande, que beaucoup considèrent comme un mets délicat. Ils sont également les mammifères les plus trafiqués aujourd'hui dans le commerce illégal d'espèces sauvages.
Le SRAS-CoV-2 est différent des autres coronavirus connus, avec 88 % ou moins d'identité de séquence. Sur la base d'analyses phylogénétiques, le SRAS-CoV-2 observé chez l'homme, les chauves-souris (RaTG13) et les pangolins malais est une nouvelle classe de coronavirus bêta. Près de 35 types différents de souches de coronavirus provenant de différentes parties du monde et de différents organismes ont été analysés au niveau du génome entier. Le SRAS-CoV-2, illustré en bleu ci-dessous, est une nouvelle classe de bêta-coronavirus (Figure 1).
Comment le coronavirus pénètre-t-il dans l'hôte ?
L'une des protéines du coronavirus, appelée protéine Spike, semble jouer un rôle important dans ce processus. La protéine Spike est une machine moléculaire multifonctionnelle constituée de deux sous-unités principales, S1 et S2. La protéine Spike se lie d'abord à un récepteur à la surface de la cellule hôte via sa sous-unité S1, puis fusionne les membranes virales et de l'hôte via sa sous-unité S2. Le domaine dans S1 de différents coronavirus reconnaît une variété de récepteurs de l'hôte, conduisant à l'attachement viral. Le domaine de liaison au récepteur (RBD) qui est de 193 acides aminés se lie et se connecte à la cellule hôte. Le récepteur du SRAS-CoV-2 chez l'homme est l'enzyme de conversion de l'angiotensine 2 (ACE2). L'ACE2 est fixé à la surface externe des membranes cellulaires des poumons, des artères, du cœur, des reins et des intestins. L'ACE2 abaisse la tension artérielle en catalysant le clivage de l'angiotensine II, un peptide vasoconstricteur, en angiotensine 1-7, un vasodilatateur. Malheureusement, ACE2 semble également être un point d'entrée populaire pour les coronavirus.
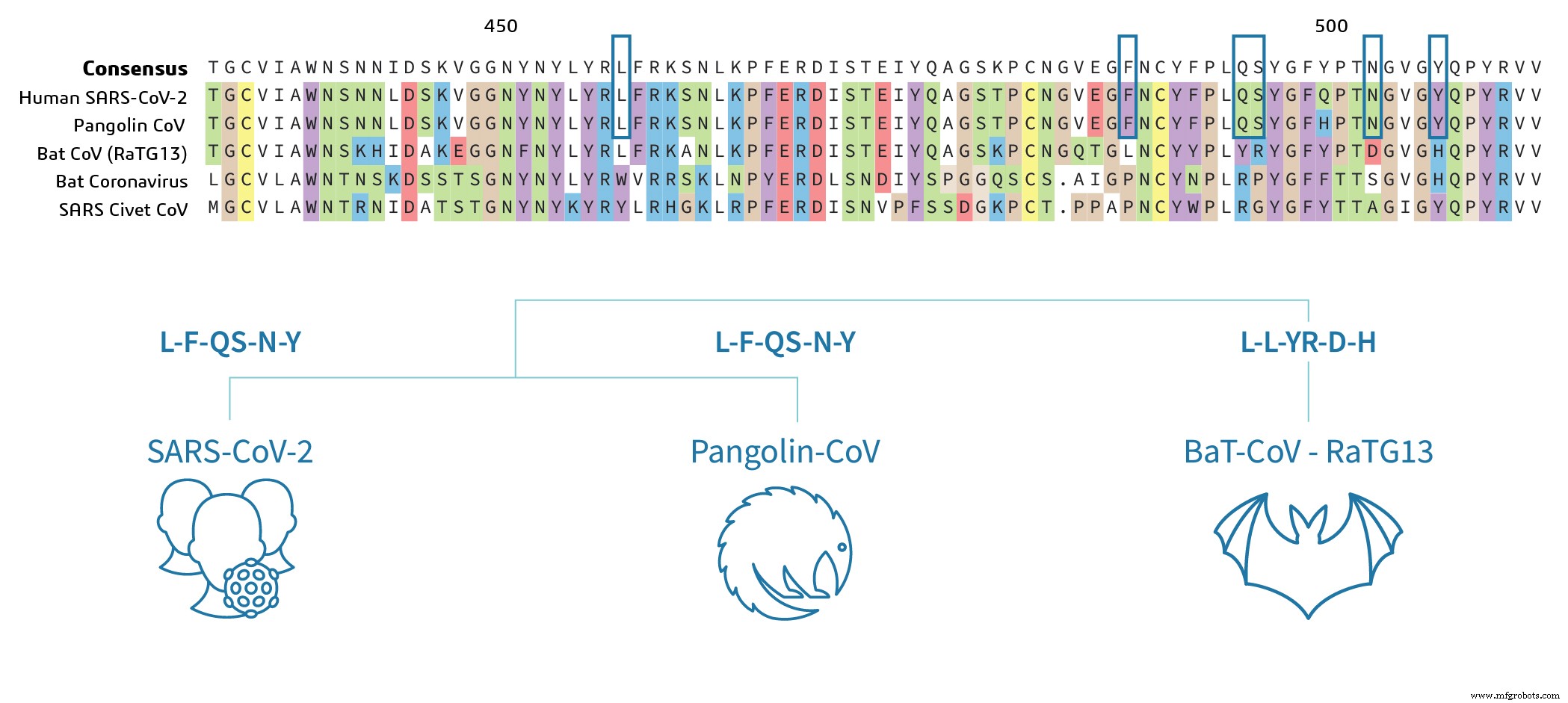
Les séquences du Pangolin CoV et du SARS-CoV-2 sont hautement conservées dans la région RBD, ce qui indique que le potentiel pathogène du virus est très similaire entre le Pangolin CoV et le SARS-CoV-2. Les résidus d'acides aminés clés, qui déterminent la liaison, sont identiques entre le Pangolin CoV et le SRAS-CoV-2 dans l'alignement des séquences (marqués de cases bleues sur la figure 2a) et les acides aminés clés (LFQSNY) affichés au-dessus des dessins animés sur la figure 2b . Fait intéressant, la chauve-souris SARS-CoV-2 RBD diffère par 17 résidus d'acides aminés, qui comprennent cinq résidus critiques pour la liaison 3 . Sur la base de l'analyse des données de séquence, on peut supposer que le bat-SARS-CoV-2 n'a peut-être pas les résidus clés pour se lier à la protéine ACE2 de la cellule hôte pour déclencher l'infection. Cela nécessitera des expérimentations pour confirmer.
Comme indiqué précédemment, la protéine Spike contient deux domaines fonctionnels :un domaine de liaison au récepteur et un second domaine qui contient des séquences qui médient la fusion des membranes virale et cellulaire. La glycoprotéine Spike doit être clivée par des protéases cellulaires pour permettre l'exposition des séquences de fusion et est donc nécessaire pour l'entrée dans la cellule. La comparaison de la séquence du site de clivage S1/S2 de Pangolin CoV et bat-SARS-CoV-2 montre une insertion du motif de reconnaissance de la furine. Cela indique un mécanisme distinct pour l'entrée du génome viral dans le cytoplasme de l'hôte pour la réplication, comme le montre la figure 3.
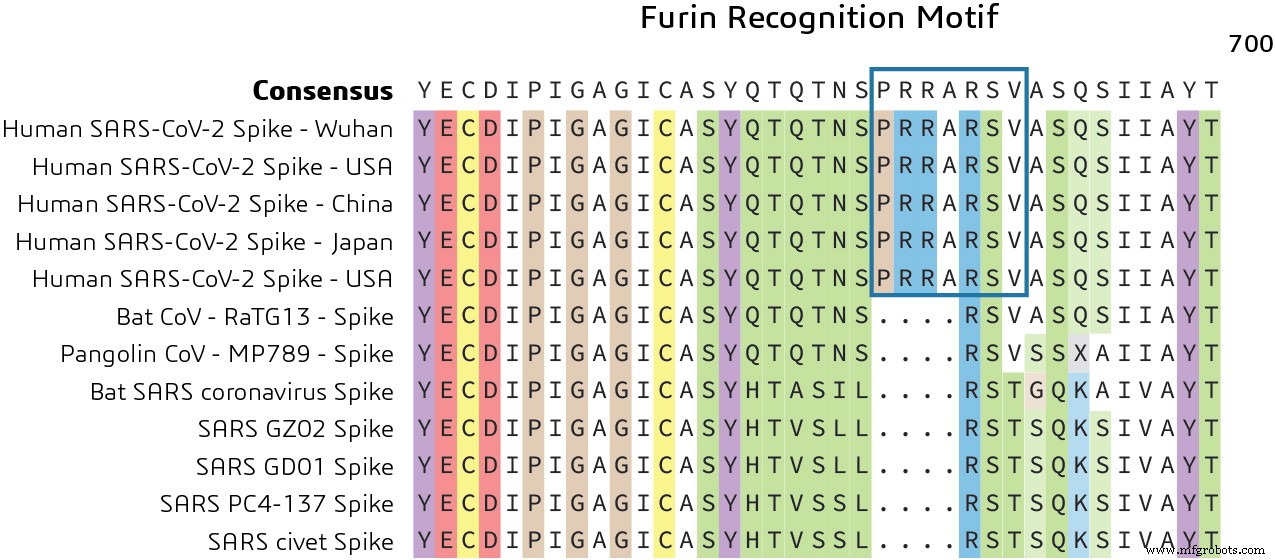
Quel est le rôle du motif de reconnaissance de la furine ? Chez l'homme, le motif de reconnaissance de la furine (PRRARSV) est reconnu par la protéine FURIN, un membre de la famille S8 des peptidases de type subtilisine qui aide à éliminer des sections de la protéine pour changer leur conformation d'un état inactif à un état actif.
Il a été suggéré que l'acquisition de ce site de clivage de la furine pourrait être un « gain de fonction » qui a permis à un CoV de chauve-souris de sauter dans l'homme et de commencer sa propagation épidémique actuelle. Cela pourrait être une piste potentielle pour explorer de nouveaux médicaments ciblant le blocage de ce motif pour empêcher la réplication du virus à l'intérieur de l'hôte.
Ainsi, un examen attentif de la protéine Spike dans le SARS-CoV-2 montre le RBD optimisé, un motif de reconnaissance de la furine, comme certains coronavirus MERS, et sa capacité à se lier fortement à la protéine ACE2. Cela suggère un processus de sélection naturelle en jeu. Il a été démontré que les événements de recombinaison naturelle dans les virus co-infectant un hôte améliorent leur gamme d'hôtes, tout en augmentant également la virulence et l'adaptation du virus. Les données du génome du SRAS-CoV-2 avec la colonne vertébrale de la chauve-souris (RaTG13) et du pangolin CoV indiquent à nouveau qu'il s'agit d'un virus généré par recombinaison naturelle .
Quel est le donneur immédiat de SARS-CoV-2 aux humains ?
La séquence SARS-CoV-2 contient un mélange à la fois de bat-SARS-CoV (RaTG13) et de régions de Pangolin CoV conservées qui ne peuvent se produire que lors de la recombinaison de ces génomes viraux. De plus, un gain de fonction, comme on le voit avec le motif de reconnaissance de la furine, implique une autre recombinaison virale. Pour que la recombinaison se produise, il est logique qu'il y ait un hôte naturel qui héberge ces génomes viraux. Est-ce un autre pangolin ? Ou un autre animal sauvage sur le marché des fruits de mer de Wuhan ? Ceci est encore inconnu. Comprendre l'origine pourrait aider à prévenir de futures épidémies de souches virales et des pandémies mondiales.
Pour plus d'informations, veuillez nous contacter :https://www.3dsbiovia.com/about/contact/.
Produits biologiques
- Le 555 IC
- La formule quadratique
- Décodage « Industrie 4.0 »
- Une approche de la chaîne d'approvisionnement pour résoudre le défi du coronavirus
- L'épidémie de coronavirus servira-t-elle de sonnette d'alarme pour les chaînes d'approvisionnement mondiales ?
- Le coronavirus détruit les chaînes d'approvisionnement traditionnelles
- Six stratégies de chaîne d'approvisionnement pour le pétrole et le gaz à l'ère du coronavirus
- Le coronavirus pourrait mettre fin aux mauvaises données d'expédition
- Comment la science des données a aidé à combattre l'épidémie de coronavirus