


Aspects théoriques du processus de réduction directe
Aspects théoriques du processus de réduction directe
Dans le procédé de réduction directe du minerai de fer, le fer métallique solide (Fe) est obtenu directement à partir du minerai de fer solide sans soumettre le minerai ou le métal à la fusion. La réduction directe peut être définie comme la réduction à l'état solide aux potentiels d'oxygène (O2) qui permettent de réduire les oxydes de fer, mais pas les autres oxydes (MnO, SiO2, etc.), aux éléments correspondants. La réduction étant à l'état solide, il y a très peu de chances que ces éléments se dissolvent (à faible activité thermodynamique) dans le fer réduit, de sorte que les oxydes qui sont plus stables que le fer restent essentiellement non réduits. La réduction directe du minerai de fer a également lieu dans la cuve du haut fourneau par les gaz ascendants.
Le système fer – oxygène
Le système fer-oxygène (Fe-O) est probablement l'un des systèmes les plus étudiés. La thermodynamique du système est bien comprise et de nombreuses informations sont disponibles sur la cinétique de réduction gazeuse impliquant les oxydes de fer. Les phases solides thermodynamiquement stables qui se produisent entre 400 deg C et 1 400 deg C dans le système Fe-O, à une pression totale de 1 kg/cm², sont représentées dans le diagramme binaire (Fig. 1). Ce diagramme montre que Fe forme avec O2 les trois composés solides stables à savoir (i) l'hématite - Fe2O3, (ii) la magnétite - Fe3O4 et la wustite - FexO, où x est un peu inférieur à 1. La phase FeO non stoechiométrique ( wustite) est instable en dessous de 570°C et se décompose en un mélange de Fe métallique et de Fe3O4. Ainsi, en lisant de droite à gauche le diagramme de phase à température constante, en dessous de 570 deg C, la séquence de phases est Fe2O3 - Fe3O4 - Fe, tandis qu'au-dessus de 570 deg C, la séquence est Fe2O3 - Fe3O4 - FeO - Fe.
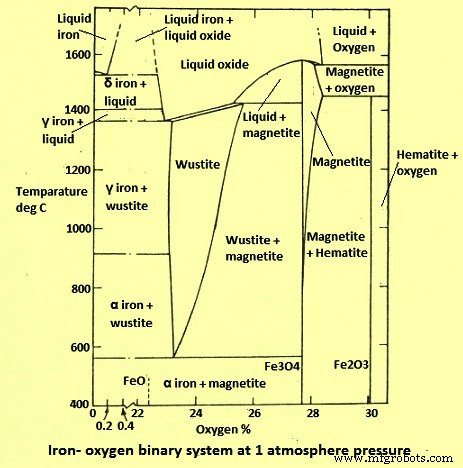
Schéma du système binaire Fe-O de la figure 1
La solubilité négligeable de O2 dans le fer alpha et gamma solide est inférieure à 0,01 % de O2. Par conséquent, la teneur en O2 n'a aucun effet sur les températures de transition des modifications solides en Fe et n'est pas prise en compte dans le diagramme.
Compte tenu de l'équilibre de la réaction, la réduction des oxydes de Fe implique une ou plusieurs de ces étapes (i) hématite (Fe2O3) -> magnétite (Fe3O4), (ii) magnétite (Fe3O4) -> fer (Fe), (iii) magnétite ( Fe3O4) -> wustite (FeO), et (iv) wustite (FeO) -> fer (Fe).
La wustite n'est stable qu'à une température supérieure à 570 deg C. Les équilibres thermodynamiques pour les réactions ci-dessus sont bien connus pour les deux principaux agents réducteurs gazeux utilisés, à savoir l'hydrogène (H2) et le monoxyde de carbone (CO).
Système Fer – Oxygène – Carbone
Les équilibres des oxydes de Fe et de Fe avec un mélange des gaz CO et CO2 (dioxyde de carbone) avec du carbone solide (C) sont illustrés à la Fig 2.
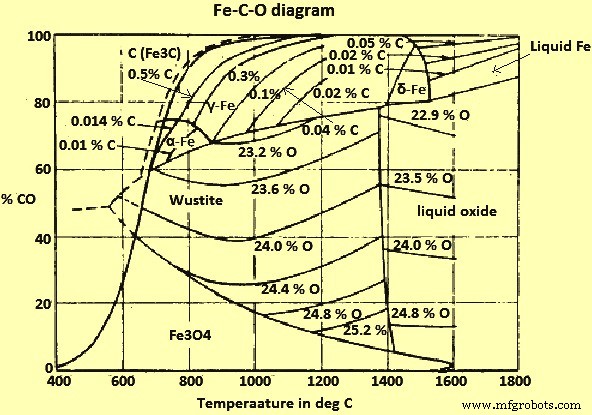
Schéma du système Fe-CO-O de la figure 2
De la figure 2, on peut déduire qu'à des températures supérieures à 710 °C et à une pression totale de 1 kg/cm², tous les oxydes de Fe peuvent être réduits par des mélanges gazeux CO/CO2 qui sont en équilibre avec C, et peuvent, par conséquent, être réduit par le C lui-même. A des températures plus basses, seuls les mélanges qui sont sursaturés en C et qui, selon l'équilibre de Boudouard, réagissent vers le dépôt de C ont une action réductrice sur la wustite.
Système fer – hydrogène – oxygène
Le diagramme d'équilibre pour les oxydes Fe et Fe avec un mélange des gaz H2 et H2O (vapeur) est illustré à la (Fig 3).
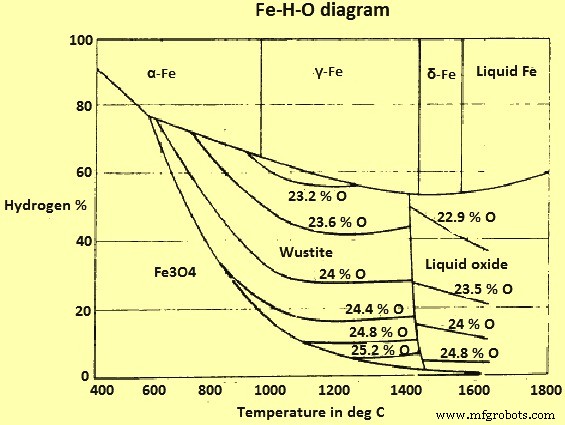
Schéma du système Fe-H-O de la figure 3
La principale différence entre ce système et le système Fe-O-C est l'absence d'une "ligne de suie" ou de phénomènes correspondants. Ainsi, il est théoriquement possible de réduire l'hématite (et la magnétite) en Fe avec H2 à n'importe quelle température.
Comparaison entre réduction par CO et H2
D'après les études des systèmes Fe-C-O et Fe-H-O (Fig 4), il semble qu'au-dessus de 815 °C, H2 soit un agent réducteur plus efficace que le CO (c'est-à-dire que les rapports d'équilibre H2/H2O sont inférieurs au CO correspondant /CO2), alors qu'il est opposé à des températures plus basses. Cependant, ces équilibres sont difficilement atteints dans les fours industriels, car la vitesse de réduction devient très lente à mesure que l'on se rapproche de l'équilibre. Lorsque les conditions s'écartent nettement de l'équilibre, les vitesses de réaction respectives pour la réduction avec H2 et CO sont dans l'ordre inverse de celles qui sont normalement attendues à partir de la considération de l'équilibre. Ainsi, H2 est en fait un agent réducteur plus efficace pour un procédé hors équilibre qui est conçu pour fonctionner à des températures inférieures à 815 deg C et le CO est plus efficace à des températures plus élevées.
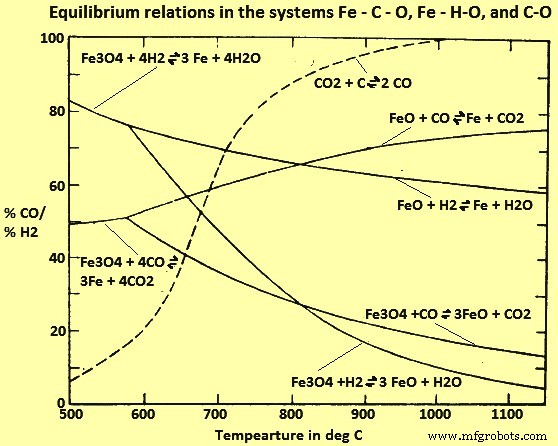
Fig 4 Relations d'équilibre dans les systèmes Fe-C-O, Fe-H-O et C-O
Des études sur l'effet du mélange de composition gazeuse de CO et H2 à différentes températures ont montré que lorsque la teneur en H2 du mélange gazeux réducteur augmente, la vitesse de réaction augmente. Cette relation s'est avérée nettement non linéaire.
La réduction des oxydes de Fe en Fe métallique avec H2 est endothermique et une source de chaleur externe est nécessaire pour maintenir la température requise. La réaction correspondante avec le CO est exothermique et dans des conditions convenablement contrôlées, la réaction est thermiquement auto-entretenue. En fait, il peut être nécessaire de diluer le CO avec du H2 ou un autre gaz absorbant la chaleur pour éviter de surchauffer la charge. Certains procédés ont été conçus pour tirer parti de l'équilibre thermique CO – H2 et utiliser des mélanges de ces gaz pour augmenter la quantité de réduction obtenue lors du chauffage du minerai, de la température ambiante jusqu'à la température maximale de réaction d'environ 1100 deg C.
La figure 4 montre que pour toutes les températures dans la plage où la réduction gazeuse est économiquement possible, les mélanges gazeux à l'équilibre contiennent au moins 60 % de CO et/ou H2. Lorsque l'équilibre n'est pas atteint, les concentrations n'ayant pas réagi de ces gaz sont encore plus élevées et la plus grande partie passe sans changement dans le four de réduction. Si le procédé doit être économique, il est alors nécessaire d'utiliser le gaz qui reste après la réduction de la wustite en Fe métallique, pour la réduction des oxydes supérieurs de Fe en wustite et/ou pour la régénération du mélange gazeux et du élimination des produits de réaction gazeux.
Réaction gaz-solide et réaction solide-solide
Les réactions gaz-solide jouent un rôle majeur dans la technologie, et englobent un domaine très large comprenant l'extraction des métaux de leurs minerais (réduction des oxydes de Fe, etc.). Une caractéristique commune à tous les systèmes de réaction gaz-solide est que le processus global peut impliquer plusieurs étapes intermédiaires. Typiquement, ces étapes intermédiaires impliquent (i) la diffusion gazeuse (transfert de masse) des réactifs et des produits de la masse de la phase gazeuse vers la surface externe de la particule solide en réaction, (ii) la diffusion des réactifs gazeux ou des produits gazeux à travers les pores de un produit de réaction solide ou à travers les pores d'un solide ayant partiellement réagi, (iii) l'adsorption des réactifs gazeux sur et la désorption des produits de réaction des surfaces solides, et (iv) la réaction chimique réelle entre le gaz adsorbé et le solide.
Dans le domaine des réactions gaz-solide, il existe plusieurs autres phénomènes qui peuvent affecter le déroulement de la réaction et les performances du four dans lequel sont réalisées les réactions gaz-solide. Ces autres phénomènes comprennent le transfert de chaleur, les modifications de la structure solide (comme le frittage) qui accompagnent la réaction, et le flux de gaz et de solides à travers le four dans lequel se produisent les réactions gaz-solide. Le taux de réduction est contrôlé par ces facteurs en fonction du procédé utilisé.
Les réactions entre solides peuvent être divisées en deux groupes principaux, à savoir (i) les vraies réactions solide-solide qui ont lieu à l'état solide entre deux particules en contact l'une avec l'autre, ou par la migration de particules à l'état solide, (par exemple la formation de carbures de fer par la réaction entre les oxydes de Fe et C), et (ii) les réactions entre les réactifs solides, qui ont lieu par le biais d'intermédiaires gazeux (par exemple, la réduction des oxydes de Fe avec du carbone à une pression de 1 kg/cm²).
La réduction des oxydes de Fe avec du C solide peut également être une véritable réaction solide-solide, à condition qu'elle soit effectuée à des pressions absolues très basses. Au cours d'une des études réalisées par la réaction de mélanges de graphite en poudre fine (C) et de minerai d'hématite sous un vide de 0,0005 mm de Hg (mercure), il a été constaté qu'à des températures allant jusqu'à 900 deg C, la réaction se déroulait très lentement, et en 18 heures seuls Fe3O4 et FeO se sont formés, mais pas de Fe. Au cours de l'essai, une transformation appréciable n'a été observée qu'à des températures plus élevées. Il a été conclu au cours de l'étude que la vitesse de réaction est déterminée par la diffusion des ions Fe dans la phase oxyde. Une déduction faite dans une autre enquête était que le C diffuse dans l'oxyde de Fe, n'a peut-être qu'un intérêt historique. Cependant, ces études ont montré que la vitesse de réaction augmente nettement lorsque la pression de gaz au-dessus du mélange pulvérulent augmente. Dans des tests de type similaire, dans lesquels un flux de N2 (azote) a été passé à travers le mélange d'oxyde de C et de Fe, une diminution marquée de la vitesse de réaction a été observée lorsque le flux de N2 a été augmenté. Toutes ces investigations, qu'elles soient menées sous vide ou sous N2, avec le taux de réduction d'oxydes de Fe en poudre similaires en CO ou H2, ont prouvé que la réaction directe à l'état solide de C et de minerai (qui est parfois considérée comme le véritable mécanisme de la véritable réduction directe) n'a aucune importance pour le déroulement du processus de réduction dans un four industriel.
Structure poreuse du fer réduit
Les essais de réductibilité sur plusieurs minerais naturels ont montré que la porosité des particules de minerai de fer est l'un des facteurs les plus importants contrôlant la réductibilité. La réductibilité exprimée comme l'inverse du temps nécessaire pour une réduction de 90 %, variait directement avec la porosité. La réductibilité relative augmente avec une augmentation de la porosité comme indiqué par l'équation "Réductibilité relative =(porosité x 0,75) + 8,0".
La réduction des oxydes de Fe donne toujours un produit de réaction poreux. La nature de l'oxyde et les conditions de réduction affectent la structure des pores du fer réduit. En effet, la réduction procède de la surface d'une particule vers l'intérieur. Le volume occupant l'espace défini par la surface de wustite d'origine est diminué. Cela ne peut être accompli qu'en développant la porosité. L'étude au microscope électronique à balayage de cette porosité a montré qu'en général, la réduction de H2 donne une structure poreuse plus fine que celle obtenue par réduction de CO. De plus, à partir des micrographies électroniques à balayage, il devient évident que la structure des pores devient plus grossière à mesure que la température de réduction de H2 augmente progressivement de 600 deg C à 1 200 deg C.
La surface de pore initiale de l'oxyde de Fe affecte la surface de pore du fer réduit formé par réduction gazeuse. Une diminution de la surface de pore initiale de l'oxyde de Fe diminue la surface de pore du fer réduit. La surface des pores du fer réduit à partir du minerai d'hématite dans H2 mesurée par la technique BET (Brunauer-Emmett-Teller) a indiqué qu'elle diminue avec l'augmentation de la température de réduction.
Une relation entre la température de réduction et la taille critique moyenne des pores et le plus petit rayon des pores a été obtenue à partir de la distribution de la taille des pores. La taille des pores s'est avérée augmenter lentement avec la température de réduction jusqu'à 900 °C, mais a augmenté rapidement avec de nouvelles augmentations de température. Ces résultats sont conformes à l'observation des surfaces de fracture par microscopie électronique à balayage, qui a montré le grossissement distinct de la structure des pores à des températures de réduction supérieures à 900 deg C.
La surface des pores du minerai d'hématite réduite est également affectée par la température de réduction et la composition du gaz. La surface des pores obtenue à partir d'hématite réduite dans un mélange gazeux CO/CO2 est d'environ les deux tiers de celle obtenue pour la réduction dans des mélanges gazeux H2/H2O. Ceci est cohérent avec la structure des pores plus grossière du fer réduit en CO/CO2 observé au microscope.
La diffusion des gaz dans les pores du fer réduit a été mesurée. Le flux diffusif dans les milieux poreux se produit via deux processus de diffusion à savoir (i) la diffusion de Knudsen, indépendante de la pression et proportionnelle à T (température) à la puissance 1/2, et (ii) la diffusion moléculaire, inversement proportionnelle à la pression et proportionnelle à T la puissance 3/2.
La structure idéale limite est supposée avoir des pores de taille uniforme qui sont tous interconnectés et se croisent avec un angle de 45 degrés.
La diffusivité effective varie pour un milieu poreux donné avec la température et la pression et est différente pour différentes paires de gaz binaires. La structure des pores devient plus fine avec la diminution de la température de réduction.
Modes de réduction
La réduction des particules de minerai de fer naturel ou des pastilles d'hématite frittées entraîne la formation de couches de produit. Ce phénomène bien connu a fait l'objet de nombreuses études. Dans l'une des études récentes de la réduction de pastilles d'hématite frittées par H2, on a remarqué qu'il existe un exemple typique de formation de couches en section polie d'une pastille d'hématite partiellement réduite. Des interfaces relativement lisses entre les couches apparaissent généralement à de faibles grossissements, bien qu'une telle apparence puisse être trompeuse.
Cela indique que la diffusion de gaz est suffisante dans la couche de wustite pour donner une certaine réduction interne en avant de l'interface Fe/FeO qui avance. La zone de réduction interne est étendue lorsque (i) la température est abaissée, (ii) la porosité est augmentée et (iii) la taille des particules devient plus petite.
L'effet de la taille des particules sur le temps nécessaire pour atteindre un pourcentage de réduction donné dépend du mode de réduction et donc du type de processus de contrôle de la vitesse. L'examen des modes de réduction des oxydes de Fe poreux par réduction gazeuse a montré trois processus de contrôle de la vitesse limitante, à savoir (i) la réduction interne uniforme, (ii) la limitation du contrôle mixte et (iii) la diffusion dans la couche poreuse de Fe. Si la réduction est contrôlée uniquement par l'un d'entre eux, alors le temps de réduction est lié au diamètre de la particule (sphéroïdal) de l'une des trois manières, à savoir (i) la réduction interne uniforme, c'est-à-dire que le temps est indépendant du diamètre, (ii) limiter le contrôle mixte, et (iii) la diffusion dans le fer poreux.
Le processus de contrôle de la vitesse devient relativement simple uniquement lorsque (i) il y a une réduction interne uniforme, donc une petite taille de particules est nécessaire, ou (ii) le contrôle ultime de la vitesse par diffusion de gaz dans les pores de la couche de fer prédomine, puisque la taille des particules est large. Il doit également être réalisé qu'il peut y avoir une transition d'un processus de contrôle de vitesse limite à un autre au fur et à mesure que la réduction progresse, en fonction de la température, de la composition du gaz, de la taille des particules et du type d'oxyde. La réduction des oxydes de fer peut également montrer un comportement inexpliqué et inhabituel.
Taux de réduction des particules de minerai de fer poreux
La porosité et la structure des pores du minerai ont un effet marqué sur l'étendue et l'uniformité de la réduction interne. Dans l'une des études, l'effet de la taille des particules sur le taux de réduction du minerai d'hématite pour un mélange de 90 % de CO et de 10 % de C02, et pour H2 à 1 000 deg C a montré qu'avec l'augmentation de la taille des particules, la réduction interne est confinée vers les régions extérieures des particules, d'où une diminution du taux global de réduction avec l'augmentation de la taille des particules.
Aux premiers stades de la réduction des particules d'hématite poreuses, il y a une conversion rapide en FeO suivie d'une réduction interne de FeO en Fe. Dans le cas limite d'une diffusion gazeuse quasi parfaite dans les pores de l'oxyde de Fe, la réduction interne prédomine et la vitesse est contrôlée principalement par une réaction gaz-solide sur les parois des pores. Une couche de Fe, épaisse de quelques atomes, est supposée recouvrir les parois des pores de FeO. Le taux de réduction est supposé être contrôlé conjointement par la diffusion rapide d'O2 à travers le revêtement de la couche de Fe sur les parois des pores et par la réaction chimique de H2 ou CO avec l'O2 à la surface de cette très fine couche de Fe.
L'effet de la taille des particules montre que le taux de réduction augmente avec la diminution de la taille des particules. La micrographie typique indique que le mode de réduction varie d'un grain à l'autre au sein de la particule. Ceci est dû aux différences locales de porosité des grains d'oxyde. En raison des variations de la taille des pores et de la diffusion plus rapide des gaz dans les pores plus grands, la majeure partie de la réaction se produit sur les parois des pores plus grands. C'est-à-dire que seule une fraction de la surface totale des pores est censée être utilisée pour la réaction. Le taux de réduction de H2 à 800 °C obtenu avec divers types de particules de minerai d'hématite augmente de manière non linéaire avec l'augmentation de la surface des pores du Fe (ou FeO) formé. Ces résultats confirment le fait que plus la surface des pores est grande, plus la fraction de la paroi totale des pores utilisée dans la réaction est petite.
Le taux de réduction interne dans les mélanges gazeux H2 - CO est généralement la somme des deux taux de réduction individuels avec H2 et CO. Les données de réduction et les dépôts de C observés indiquent qu'en dessous de 1000 ° C, les réactions gazeuses conduisant à l'eau- l'équilibre des gaz est lent.
Taux de réduction du minerai de fer (morceau ou boulette)
Le taux de réduction du minerai en morceaux ou des boulettes de minerai est de nature complexe dans le flux de gaz réducteur dans un lit à garnissage, La complexité est due au fait que le taux global de réduction est contrôlé par plusieurs processus de réaction en série, tels que la chaleur et la masse transfert à travers la couche limite du film de gaz, réactions gaz-solide et diffusion de gaz dans les couches de produit poreux. Grâce à des analyses mathématiques, facilitées par des calculs informatiques, de nombreuses équations ont été dérivées pour décrire le taux de réduction des grosses particules d'oxyde pour différents modes de réduction.
Dans plusieurs expériences avec des pastilles simples ou des particules de minerai de fer, le transfert de chaleur est relativement rapide et, avec des flux de gaz à vitesse suffisamment élevée, la résistance au transfert de masse du film gazeux est suffisamment faible pour être négligée. Par conséquent, il existe principalement deux étapes de réaction majeures en série qui influencent la vitesse de réduction, à savoir (i) les réactions gaz-oxyde et (ii) la diffusion de gaz dans les couches poreuses d'oxyde et de produit poreux. Les effets relatifs de ces processus de vitesse dépendent de la taille des particules, de la composition du gaz, de la température et du mode de réduction, et ils changent avec la progression de la réduction.
Diffusion de gaz dans la couche poreuse de Fe
Dans l'une des études, des expériences de réduction unidirectionnelle ont été réalisées pour démontrer l'effet de la diffusion de gaz dans les pores des couches de Fe. De longs échantillons de cylindres ont été préparés à partir de gros morceaux de minerai d'hématite en morceaux et ont été emballés à l'intérieur d'un tube de nickel étroitement ajusté. Après réduction de H2 pendant le temps nécessaire, l'échantillon a été partitionné axialement et poli, et l'épaisseur de la couche de Fe a été déterminée. Les résultats des expériences ont montré que, lorsque l'épaisseur de la couche de Fe réduite était d'environ 1 mm, la réduction supplémentaire se déroule en accord avec la loi de vitesse parabolique, ce qui est similaire au résultat du contrôle de la diffusion des pores. Ces tests ont démontré qu'à mesure que l'épaisseur de la couche poreuse de Fe augmente, le taux de réduction est finalement contrôlé par la diffusion de gaz dans les pores de la couche de Fe.
Une réduction interne partielle précédant le front d'avancement principal de la couche de Fe peut conduire au piégeage d'une partie de FeO dans la couche réduite. Cette situation peut entraîner une élimination lente de l'O2 dans les dernières étapes de la réduction.
Au fur et à mesure que la température de réduction diminue, la structure des pores devient beaucoup plus fine, vraisemblablement avec de nombreux canaux étroits et des goulots d'étranglement sur les capillaires connectés, lorsque la diffusion de Knudsen prédomine, d'où de faibles valeurs du rapport de la diffusivité moléculaire effective/diffusivité moyenne effective de Knudsen. Au fur et à mesure que la structure des pores devient plus grossière avec l'augmentation de la température de réduction, ce qui facilite le passage du gaz à travers les pores, le rapport devient plus élevé.
L'effet de la composition du gaz sur le temps nécessaire pour atteindre 50 %, 75 %, 90 % et 95 % de réduction pour les boulettes de minerai d'hématite frittées et les boulettes de minerai de magnétite réduites à 900 deg C par des mélanges H2-CO-CO2 (avec un rapport CO/CO2 égal à 9 pour supprimer le dépôt de suie), est que lorsque H2 est remplacé par du CO, le temps de réduction isotherme pour atteindre un pourcentage donné d'élimination d'O2 augmente progressivement jusqu'à environ 50 % de CO et avec un ajout supplémentaire de CO, il y a une augmentation marquée de le temps de la réduction. Le temps de réduction pour 100 % (rapport CO/CO2 égal à 9), est environ 10 fois plus long qu'en H2 à la même température. La diffusivité moléculaire des gaz dans un système binaire, tel que H2-H2O ou CO-CO2, telle que dérivée de la théorie cinétique des gaz, est un invariant pour le système et essentiellement indépendant de la composition du gaz. Cependant, dans les systèmes ternaires et multi-composants, chaque espèce a une diffusivité différente et varie avec la composition du gaz. De plus, l'équation de vitesse pour le flux diffusif est compliquée.
Le comportement de réduction des boulettes de minerai d'hématite dans les mélanges H2-CO a montré un schéma similaire à celui observé dans le H2 et le CO, c'est-à-dire que le taux de réduction au-delà d'environ 50 % d'élimination d'O2 est contrôlé par la diffusion de gaz dans les pores du Fe calque.
Limitation du contrôle mixte dans le débit initial
Dans les premiers stades de la réduction, la vitesse de réduction est contrôlée conjointement par (i) la diffusion de gaz dans les pores du FeO (la diffusion à l'état solide dans FeO peut être ignorée) et (ii) la réaction sur les parois des pores du FeO . Cela implique une fine couche poreuse de Fe et une diffusion rapide des gaz dans celle-ci. En fonction de la porosité de FeO et de la diffusivité du gaz dans celui-ci, il y a une réduction interne partielle en avant de l'interface Fe/FeO nominale. La réaction de H2 avec FeO poreux est généralement confinée aux bouches des pores proches de l'interface nominale Fe/FeO.
Réduction interne partielle
En fonction de la composition du gaz, de la température, de la taille des granulés et de la pression totale du gaz, il y a un contrôle de débit mixte pendant une certaine période de la réduction dans le cadre des lois de débit limite. L'équation de vitesse est normalement basée sur l'hypothèse que la réduction gazeuse de la pastille est contrôlée conjointement par une lente diffusion à contre-courant du gaz à travers les pores inter-particules de la pastille, et par une réaction chimique lente du gaz avec l'oxyde de Fe à la Interface oxyde de Fe-/Fe des particules.
Réaction de déplacement eau-gaz
La réaction de water gas shift joue un rôle important dans les procédés de réduction directe qui utilisent des hydrocarbures reformés comme réducteur dans la réduction des oxydes de fer. Il est généralement admis, d'après les différents taux de réduction du minerai de fer par le CO et par le H2 et l'effet marqué que même de faibles quantités de H2 contenu dans un mélange CO/CO2 ont sur le taux de réduction, que le H2 est le véritable composant réducteur dans de tels mélanges gazeux. Le CO est considéré comme servant principalement à réduire la vapeur résultante (H2O) en H2. Les réactions sont (i) H2 + FeO =H2O + Fe, et (ii) H2O + CO =H2 + CO2.
Le deuxième sous-processus de cette réaction est connu sous le nom de réaction de conversion eau-gaz. Il est bien connu que ce processus a besoin d'un catalyseur. Dans la réduction du minerai de fer, tous les produits (Fe3O4, FeO et Fe) sont considérés comme des catalyseurs possibles. Parmi ceux-ci, le Fe solide est particulièrement actif. Le processus de réduction du minerai de fer dans des mélanges CO/CO2 contenant H2 doit donc être compris, lorsque Fe métallique est présent, comme une séquence de réaction. La sous-réaction (i), la réduction proprement dite, a lieu à la surface de l'oxyde de Fe tandis que la sous-réaction (ii), la régénération de l'H2 par la réaction eau-gaz, a lieu à la surface du Fe.
La séparation spatiale des deux sous-réactions nécessite leur connexion par un processus de transport, qui doit avoir lieu sous la forme d'une diffusion gazeuse ou d'une diffusion de surface par l'un des participants à la réaction. Les conditions optimales se produisent à la limite triphasée Fe/Fe oxyde/gaz.
Gonflement pendant la réduction
Le volume apparent de minerai de fer ou de boulette augmente généralement pendant la réduction. C'est ce qu'on appelle un gonflement. Il existe globalement trois types de comportement de gonflement qui peuvent être observés. Ceux-ci sont connus sous le nom de (i) gonflement normal, (ii) gonflement catastrophique dans lequel il y a une expansion de volume soudaine avec la conversion de FeO en Fe, le Fe apparaissant sous la forme d'excroissances filamenteuses, appelées moustaches de Fe fibreux, et (iii) l'expansion par éclatement, un comportement typique des matériaux riches en fer contenant de petites quantités d'alcalis. Ce dernier type de comportement est différent du gonflement catastrophique (bien qu'il n'en soit pas moins grave) en ce sens qu'une grande partie de l'expansion a lieu avant l'apparition de Fe comme produit de réaction.
On peut dire que ni le minerai en morceaux ni l'aggloméré ne sont connus pour gonfler de manière anormale ou catastrophique, alors que certains types de boulettes le font, et donnent lieu à des problèmes opérationnels en réduisant la perméabilité de la charge car les boulettes anormalement gonflées sont molles, spongieuses et ont tendance à se désintégrer. .
Les volumes spécifiques de différents oxydes de Fe et de Fe tels que rapportés dans la littérature sont de 0,272 cc de Fe2O3 par gramme de Fe (à température ambiante), 0,270 cc de Fe3O4 par gramme de Fe, 0,231 cc de FeO par gramme de Fe (23,5 % O2) , et 0,128 cc de Fe par gramme de Fe. Par conséquent, le volume devrait diminuer à chaque étape de réduction. Cependant, la principale cause de gonflement des minerais de Fe est causée par la transformation du minerai d'hématite hexagonale en minerai de magnétite cubique et les perturbations du réseau qui en résultent. Les perturbations du réseau provoquent la formation de pores, ce qui entraîne une augmentation considérable du volume apparent des minerais de fer lors de la transformation de l'hématite en magnétite.
En général, lors de la réduction dans un gaz riche en CO, le gonflement est beaucoup plus important que dans un gaz riche en H2. La raison de ce comportement est que la poussière métallique se produit lors du dépôt de C dans des mélanges gazeux contenant du CO. Cependant, il est difficile d'expliquer le gonflement qui peut se produire lors de la réduction des mélanges gazeux CO-CO2 lorsqu'il n'y a pas de dépôt de C. La cause et l'effet du gonflement ou du rétrécissement accompagnant la réduction n'ont pas encore été résolus.
Il existe généralement deux types d'impuretés dans les boulettes de minerai. Il s'agit (i) des impuretés ayant un effet gênant sur le gonflement, et (ii) des impuretés ayant un effet améliorant le gonflement. L'exemple du premier est la silice (SiO2) tandis que pour le second les alcalis (K2O, Na2O). Il a été remarqué que les pastilles de Fe2O3 de qualité réactif contenant du SiO2 jusqu'à 5 % ne gonflent pas lorsqu'elles sont réduites en mélanges gazeux CO - CO2 et qu'une certaine quantité de SiO2 est également nécessaire dans les pastilles acides pour maintenir leur résistance et éviter un gonflement catastrophique. Dans le second cas, on voit que de petites additions d'alcalis Na2CO3 ou K2CO3 dans la plage de 0,1 % à 1 % peuvent entraîner le gonflement catastrophique en H2 ou CO de pastilles de minerai autrement normales. L'effet des alcalis devient plus prononcé avec l'augmentation du rapport de basicité (CaO/SiO2) dans la pastille. L'effet indésirable peut être évité par l'ajout d'une gangue acide à grains fins pour former des silicates alcalins stables.
Il existe des observations contradictoires sur l'effet des impuretés dans les boulettes de minerai (par exemple, la teneur en chaux). Une petite quantité d'ajout de CaO (moins de 0,1 %) aux boulettes de minerai d'hématite provoque un gonflement considérable pendant la réduction, ce qui suggère que CaO est une cause de gonflement catastrophique. D'autre part, il a été remarqué qu'environ 1 % d'ajout de CaO aux boulettes de minerai d'hématite supprime le gonflement pendant la réduction. Ces variations de l'effet observé du CaO sur le gonflement peuvent être dues à la présence ou à l'absence d'autres impuretés dans le minerai de fer, telles que les alcalis.
Réduction du minerai d'hématite par C
La réaction entre le minerai d'hématite et le C est d'une importance fondamentale dans la préparation de boulettes de minerai métallisé. Une grande partie du nouvel intérêt a été stimulée par le développement du procédé du four rotatif qui utilise du C solide comme réducteur dans la production de fer à réduction directe (DRI). Il est généralement admis que la réduction de l'oxyde de Fe par C se produit à travers les intermédiaires gazeux CO et CO2, sauf sous un vide très poussé où la véritable réaction solide-solide est le mécanisme prédominant.
Le mécanisme de réaction par intermédiaires gazeux qui a lieu lors de la réduction du minerai d'hématite par C se fait par des réactions (i) C(s) + 0,5 O2 =CO(g), (ii) FexOy(s) + CO(g) =FexO (y-1) (s) + CO2(g), et (iii) CO2(g) + C(s) =2CO(g).
La formation initiale de CO est une étape importante dans la vitesse de réaction globale. L'O2 de l'air piégé avec le gaz O2 libéré par la dissociation des oxydes de Fe réagit avec le C pour donner du CO (première réaction). De plus, du CO peut également être formé par une véritable réduction directe se produisant aux points de contact entre les particules d'oxyde de C et de Fe. Le gaz CO ainsi produit réagit facilement avec les particules de minerai d'hématite (seconde réaction). La réaction de Boudouard ou de perte de solution entre le gaz CO2 et les particules C régénère le gaz CO (troisième réaction) et tend ainsi à restaurer le potentiel réducteur de la phase gazeuse contenue dans les pores de l'échantillon. L'oxydation de certains types de C en CO2 est catalysée en présence de certains métaux et composés métalliques. L'amélioration de la vitesse du processus a été observée avec l'ajout de Li2O (oxyde de lithium) et l'effet inhibiteur a été rapporté avec l'ajout de FeS (sulfure ferreux). Le Fe métallique s'est avéré être un bon catalyseur pour la gazéification du graphite (C). En raison de cette réaction catalytique imprévisible dans le mélange, les équations dérivées par modélisation mathématique pour décrire la vitesse globale de la réaction ont une valeur limitée et ne peuvent s'appliquer qu'aux systèmes où les réactions ne sont pas catalysées.
À des températures modérément élevées (par exemple 1000 deg C), les vitesses des réactions d'oxyde de Fe (à une température supérieure à 570 deg C et la séquence est Fe2O3, Fe3O4, FeO, Fe) sont bien supérieures à celles de la réaction de Boudouard. En d'autres termes, le processus global devient limité par la disponibilité du gaz CO selon la réaction de Boudouard. Thus at steady-state the composition of this gas-phase closely corresponds to the equilibrium gas-phase composition for FexOy/FexO(y-1).
Fe oxides reduction with hydrocarbons
Hydrocarbons can be used in two ways as a reducing agent for the production of DRI. These are (i) direct use of hydro-carbons or a mixture of gas containing hydro-carbons, and (ii) use of the reformed hydrocarbon products (CO, H2), by reforming within the reduction reactor (it has been found that auto-catalytic reforming of some hydro-carbons within the reducing furnace provided an access of macro and micro porosity which leads to more extensive reduction and also which leads to the deletion of the capital cost of gas reformer and processing.
There are a few studies using directly hydrocarbons or a mixture of gas containing hydrocarbons as reductant for direct reduction of iron ores. Two important points emerge from these studies. The first is that the rate of reduction with hydrocarbons is slow and the production of a high quality of DRI is troublesome and uneconomical. The second point is that these studies have been done under isothermal conditions in a thermo-gravimeter with single particle or powder compact, thus the results are of only theoretical value.
Theoretical importance of investigations with hydrocarbons – The kinetics of ferric oxide reduction by pure methane (CH4) has been studied in the three temperature ranges of (i) low temperature (500 deg C to 600 deg C), (ii) medium temperature (650 deg C to 750 deg C) and (iii) high temperature (800 deg C to 950 deg C). At the low temperature, the reduction proceeds only from Fe2O3 to Fe3O4. A prolonged holding of the sample in a stream of CH4 has not led to any process extension beyond this stage. The rate became appreciable at 650 deg C. In special experiments after the Fe3O4 composition has been reached, the sample has been reduced further by H2 and CH4. It has been shown that CH4 reduction in the low temperature range beyond the Fe3O4 stage occurs only if a sufficient quantity of metallic Fe has been built up. In this case the reducing agent has not been CH4, but its decomposition product, H2. C formed by CH4 decomposition takes almost no part in the reduction and gets accumulated in the sample.
In the medium temperature range the conversion of Fe3O4 to FeO takes place but at low rates. A sharp rise in reduction rate is observed on going from 750 deg C to 800 deg C. The process becomes very sensitive to temperature changes beyond 800 deg C, and accelerated considerably in the high temperature range, when metallic Fe appeared in the sample. The appearance of metallic Fe at the FeO to Fe stage, at comparatively high temperatures indicates a decisive role of metallic Fe as a catalyst for reforming CH4 by the reduction products (CO2, and H2O). In the absence of a catalyst, the decomposition of CH4 and its reforming by the reduction products (CO2, H2O) do not occur to any substantial extent and no C accumulation in the sample has been observed. When the Fe catalyst is present, CH4 dissociation into the elements takes place only at very late stages of reduction, when there is insufficient CO2 and water vapour to convert all the CH4 diffused into the sample. C build-up in the sample starts from that stage.
In the 2-stage production of DRI with CH4, it has been found that the complete decomposition of CH4 in the presence of the Fe bearing material occurs at temperatures of 850 deg C to 900 deg C, which is 400 deg C to 450 deg C lower than on an inert surface (e.g. fire clay), while the reaction rate, conversely, has been 10 times higher. The products of the first stage are a sooty Fe containing 30 % to 50 % C and technically pure H2.
In the second stage, the product of the first stage (sooty Fe with highly dispersed C in the pores of DRI and on the surface of the Fe particles) has been used as an active reducing agent and mixed with mill scale or concentrate. The mixture has been reduced in the temperature range 1050 deg C to 1100 deg C with a make-up reducing agent of H2 reformed natural gas. The results of industrial trials has shown that the use of sooty Fe instead of soot, petroleum coke and the other known carbonaceous reducing agents considerably intensified the Fe-oxide reduction process. As is well known, the direct reduction of Fe oxides with C is directly related to the rate of reaction between the C and CO2. The sooty Fe can have intensified the rate of Boudouard reaction.
The isothermal reduction of hematite ore pellets (with 10 % to 15 % porosity) in a thermo-balance with a mixture of CH4-H2 (containing 4.5 % CH4) within the temperature range 700 deg C to 1000 deg C has shown that the reduction is chemical – controlled initially and diffusion – controlled in the later stages. It has been shown that reduction in pure H2 is faster than in the CH4- H2 mixture. This difference is attributed to C deposition in the outer reduced layers of the pellet, causing resistance to gas diffusion when the reducing gas contained CH4. It has been shown that the excess residual C can be removed from the reduced iron at lower temperature by its hydrogenation.
In another study, it has also been demonstrated that it is possible to hydrogenate residual C in direct reduced products to CH4. The C formed as a result of the reduction of Fe oxide in a mixture of CH4 and H2 (containing 20 % CH4) reacted with steam (H2O) according to the water gas reaction to regenerate H2 and produce CO.
Pure ferric oxide briquettes were reduced at temperatures ranging from 800 deg C to 1050 deg C, in gas mixtures containing H2, CO, CH4, N2 and CO2, which has been obtained by partial oxidation of natural gas with air. The CH4 content of the reformed gas mixture was between 13 % and 16 %. The overall reduction rate again has been controlled initially by chemical reaction and the gaseous diffusion has been applicable during the latter stages. It has been shown that the hematite ore briquettes have swelled and considerable porosity has been was developed during reduction. The solid-state diffusion rates increased more rapidly with temperature than it did by interfacial or gaseous diffusion reaction rates. The reduction of porous (30 % porosity) Fe ore in CH4 has indicated that the reaction proceeded stepwise from Fe2O3 to Fe3O4, FeO and Fe. The Fe catalyzed the CH4 cracking reaction. Optimum conditions for CH4 utilization occurred at around 1000 deg C.
The above findings are not consistent with the earlier studies on the understanding of high-grade porous (around 30 %) or dense hematite ore reduction kinetics, which had shown that the rate of reduction can be considered to fall between 3 limiting cases, namely (i) uniform internal reduction, (ii) limiting mixed control, and (iii) diffusion in porous iron layer, respectively with the rate of reduction corresponding to, (i) chemical control, (ii) the overall chemical control and diffusion control, and (iii) diffusion control. The overall rate of reduction is not controlled by only one of these rate controlling mechanisms and can be changed from one limiting case to another during the course of reduction.
In one of the studies it has been found that the most important factors controlling the extent of reduction are (i) the temperature, (ii) the composition of gas, presence of unreacted hydrocarbons in the reducing gas, the ratio of H2/C in it, and reducing capacity, (iii) the ore particle size, and (iv) the residence time for reduction.
Reduction of Fe oxides with the products of CH4 reformed with H, O within the reduction furnace – In early 1981 a commercial process has been introduced, using gaseous mixtures containing upto around 30 % by volume of CH4 (e.g. coke oven gas), for the direct gaseous reduction of Fe ore in a counter current moving bed shaft furnace. The furnace contained a reduction zone, a cooling zone, and an intermediate reforming zone. A hot mixture of coke oven gas and steam has been fed to the intermediate zone and reduced Fe ore therein catalyzed the reforming of the CH4 to CO and H2. The reformed gas flows upward into the reduction zone for the reduction of Fe ore.
Processus de fabrication
- Processus de production d'alliage de fer et de tungstène-nickel
- Introduction au frittage laser direct des métaux
- Fer à réduction directe et ses procédés de production
- Le processus de frittage des fines de minerai de fer
- Procédé Finex pour la production de fonte liquide
- ITmk 3 Processus de fabrication de pépites de fer
- Avantages des fournisseurs de fonte en tant que processus de fabrication
- Diverses méthodes de processus de fabrication de l'acier
- Comprendre le processus de fusion du minerai de fer