


Dépistage virtuel guidé par pharmacophore pour la réaffectation de médicaments
La réorientation des médicaments est une stratégie établie de longue date pour identifier de nouvelles demandes de thérapies approuvées ou expérimentales qui dépassent le cadre de l'indication médicale d'origine. 1 Depuis le repositionnement fortuit d'un médicament conçu pour l'hypertension et l'angine de poitrine pour devenir la tristement célèbre "petite pilule bleue" de Pfizer, communément appelée Viagra, la réorientation des médicaments n'a pas été aussi importante aux yeux du public. L'urgence mondiale pour les thérapies pour traiter le COVID-19 a une fois de plus attiré l'attention des masses sur cette stratégie médicamenteuse.
La modélisation 3D des protéines comprend plusieurs méthodes de modélisation et de simulation moléculaires pour la réorientation des médicaments. Un article précédent de cette série a exploré l'amarrage moléculaire comme méthode d'identification des inhibiteurs possibles se liant à la protéase principale du SRAS-CoV-2. Est-ce qu'une alternative in silico une méthode telle que la modélisation pharmacophore obtient des résultats comparables et fournit des preuves supplémentaires à l'appui de la sélection d'un candidat plutôt qu'un autre ?
Prendre un itinéraire alternatif
La modélisation des pharmacophores fournit une abstraction des caractéristiques moléculaires nécessaires à la reconnaissance d'un ligand par une protéine cible. Sa représentation des interactions moléculaires et de la liaison offre une perspective contrastée avec les méthodes de simulation classiques.
Nous avons obtenu un ensemble de données initial de plusieurs structures de protéines de protéase principales du SRAS-CoV-2 à partir de Diamond Light Source. 2 Nous avons aligné ces structures, puis utilisé le protocole « Interaction Pharmacophore Generation » disponible dans BIOVIA Discovery Studio pour générer des pharmacophores représentant les interactions sans liaison de chaque complexe récepteur-ligand. Le nombre total de caractéristiques dans les pharmacophores allait de deux caractéristiques pour les structures cristallines 5R80 et 5R7Y à un modèle à neuf caractéristiques pour 6LU7. Nous avons fusionné les pharmacophores des complexes individuels en un seul modèle et édité des caractéristiques étroitement groupées. Le modèle final comprenait 14 caractéristiques qui représentent les contacts intermoléculaires entre la protéase et d'éventuels liants à petites molécules.
Nous avons ensuite effectué in silico mutagenèse par balayage d'alanine sur les résidus du site actif de tous les complexes pour identifier les résidus qui réduisaient l'affinité de liaison (point chaud) de ce complexe protéine-ligand lors de la mutation. De tous les complexes, nous avons identifié huit résidus (HIS41, MET49, ASN142, HIS163, MET165, PRO168, GLN189 et GLN192) comme points chauds dans au moins un complexe et trois résidus qui étaient des points chauds dans au moins quatre complexes. Dans le modèle de pharmacophore à 14 caractéristiques, six caractéristiques correspondaient à des interactions avec l'un des huit résidus de hotspot. Un deuxième jeu de données de M PRO non covalent complexes de ligands libérés par Diamond Light Source 2 a révélé un certain nombre de modes de liaison différents, nous conduisant à séparer les six caractéristiques pharmacophores clés en deux groupes à utiliser dans la prochaine étape du criblage virtuel.
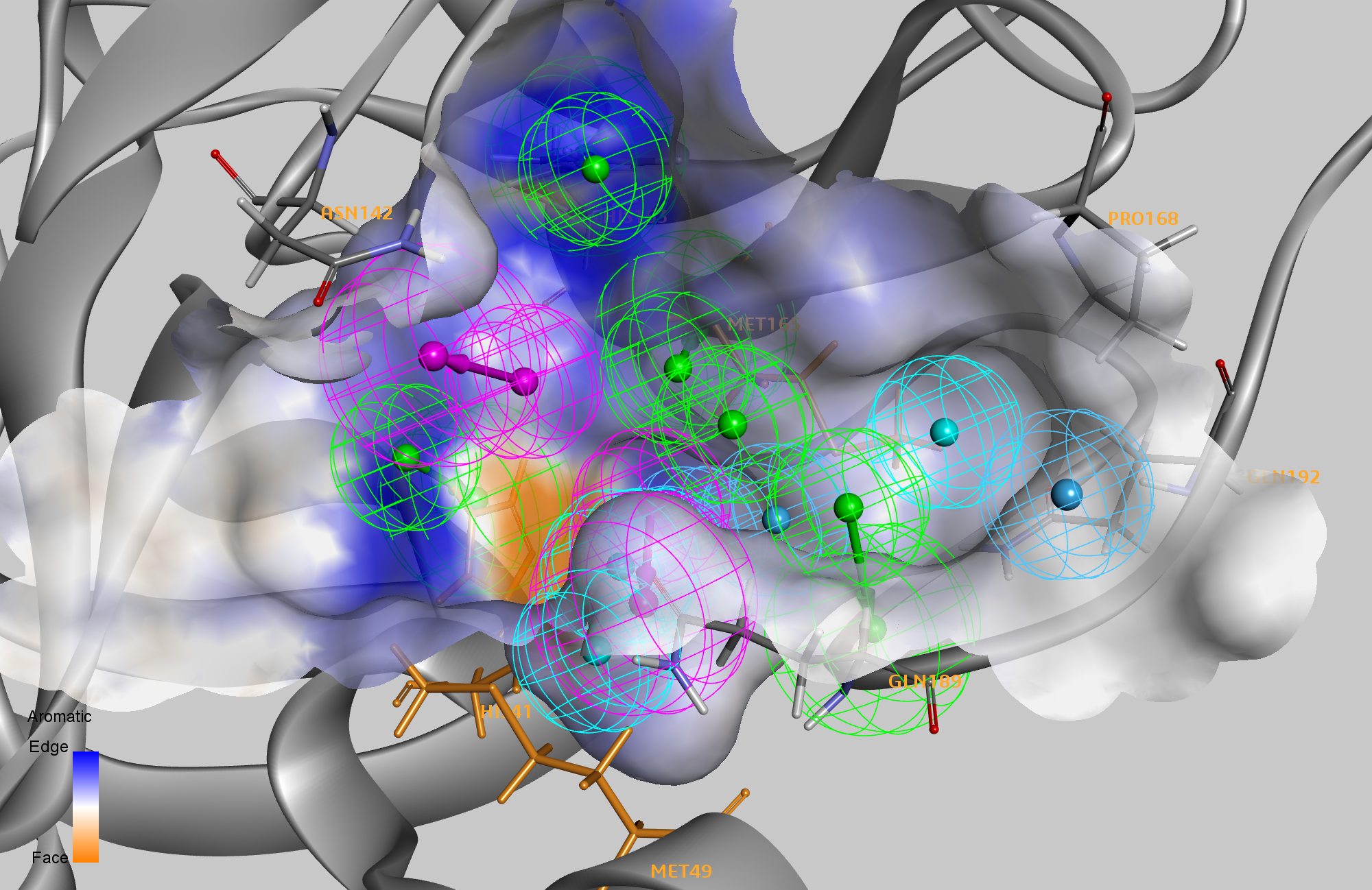
Nous avons maintenant utilisé ce modèle de pharmacophore pour effectuer un criblage virtuel d'une bibliothèque sélectionnée contenant 2 650 médicaments approuvés par la FDA qui ont plusieurs confirmations de ligands pré-générées pour une recherche rapide. Nous avons criblé ces ligands par rapport au pharmacophore à 14 caractéristiques, avec l'exigence qu'au moins une caractéristique de chacun des deux groupes requis corresponde, ainsi qu'au moins deux caractéristiques des huit autres caractéristiques, pour identifier un hit. Il s'agit de permettre une exploration diversifiée des différents modes de liaison des ligands, tout en garantissant que seules les poses pouvant former au moins deux interactions avec des résidus significatifs identifiés dans la mutagenèse par balayage d'alanine sont conservées. L'algorithme explore essentiellement plus de 12 000 combinaisons possibles de pharmacophores. Nous avons ensuite minimisé les meilleurs résultats d'ajustement pour chaque combinaison de pharmacophores au sein de la protéine. Enfin, nous avons déterminé les énergies de liaison des poses optimisées dérivées du pharmacophore à l'aide de CHARMm avec un modèle de solvant implicite généralisé Born utilisant le volume moléculaire (GBMV). Cette méthode est effectivement un calcul d'amarrage moléculaire (comme dans notre blog précédent), mais ici nous avons utilisé notre modèle de pharmacophore optimisé pour restreindre et filtrer les résultats amarrés.
Nous avons calculé les interactions non liées de chaque pose et filtré pour nous concentrer uniquement sur les poses avec des interactions avec quatre résidus clés - HIS41, MET49, CYS145 et MET165. HIS41, MET49 et MET165 étaient les trois résidus communs à au moins quatre complexes identifiés précédemment, et CYS145 est le deuxième résidu de la dyade catalytique significative HIS41/CYS145 trouvée dans chaque sous-unité de l'homodimère de protéase principal. Nous avons trié les résultats avec l'énergie libre de liaison calculée pour identifier les dix meilleurs candidats plausibles pour une exploration plus approfondie. Le ritonavir était le seul ligand commun identifié ici et dans le blog précédent. Le ritonavir avait la septième meilleure énergie libre de liaison et était également précédemment classé septième dans l'étude d'amarrage. Le ritonavir fait actuellement l'objet de plusieurs essais cliniques pour le COVID-19. 3
Vidéo 1 :Pose la mieux notée pour le Ritonavir en interaction avec HIS41, MET49, SER144, CYS145, MET165, GLU166 et GLN189.
Le meilleur ligand de notation dans la méthode présentée ici était le montélukast, un antagoniste des récepteurs des leucotriènes cystéinyliques. Un article publié récemment a émis l'hypothèse de son utilisation pour limiter la progression de la maladie sur l'infection au COVID-19. 4
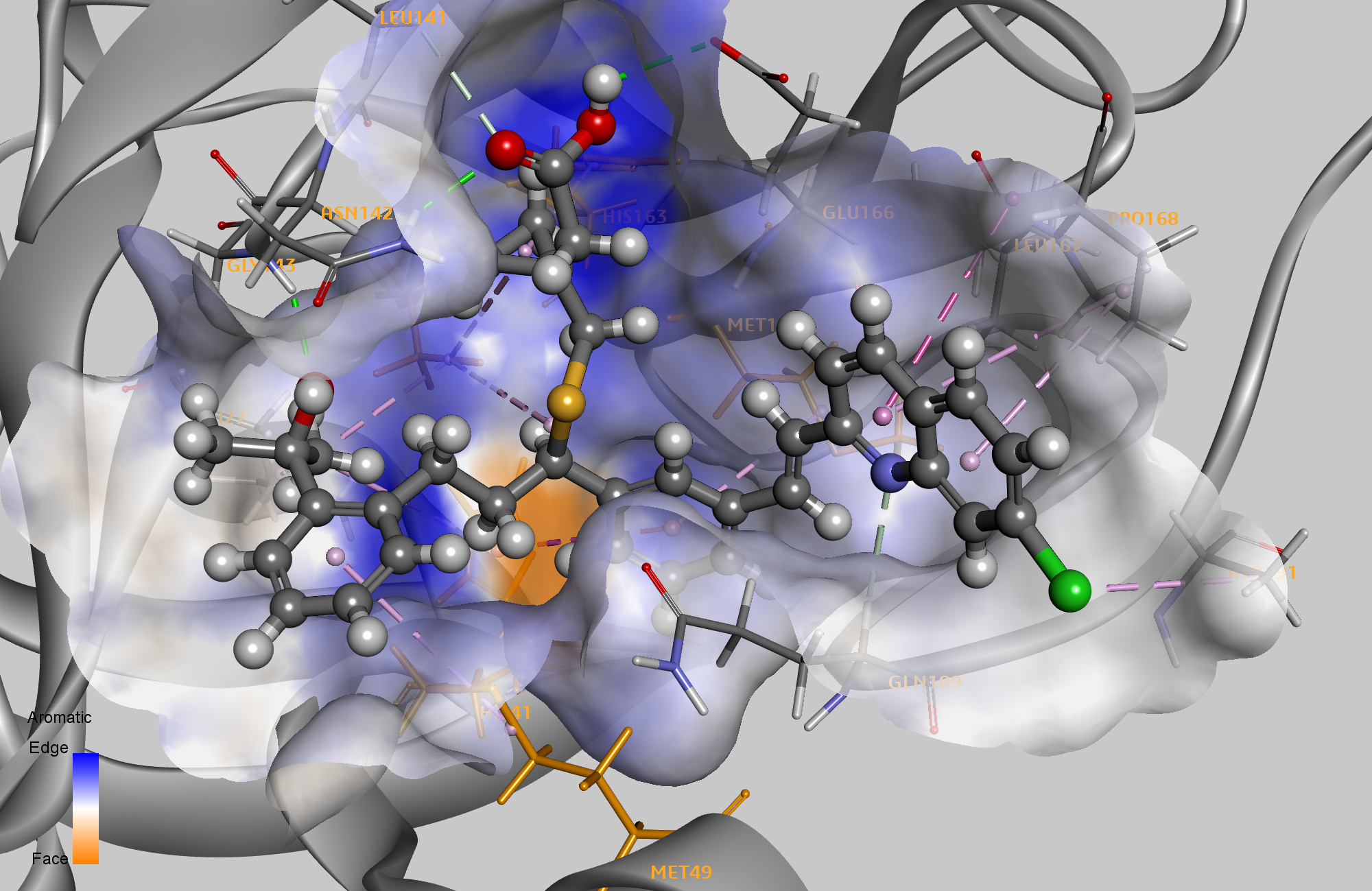
Trois autres composés les mieux notés comprenaient les médicaments Telmisartan, Moexipril et hydroxycloroquine. Le potentiel de tous ces composés en tant que médicaments réutilisés pour COVID-19 a récemment fait l'objet d'un examen minutieux, 5 l'hydroxychloroquine suscitant le plus de controverses. Une différence supplémentaire entre les dix premières listes de résultats des deux méthodes est la diversité des cibles médicamenteuses incluses. Les dix premiers résultats d'amarrage comprennent sept inhibiteurs de la protéase du VHC ou du VIH. La liste de résultats prioritaires par pharmacophore comprend des médicaments avec sept classes cibles différentes.
Notre criblage virtuel dérivé du pharmacophore a produit une liste de résultats prioritaires comprenant plusieurs nouveaux médicaments COVID-19 potentiels non identifiés lors de nos précédents travaux d'amarrage. Avec cette approche, nous démontrons l'utilisation de in silico la mutagenèse par balayage à l'alanine en tant que technique utile pour affiner le modèle de pharmacophore. Nous avons également imposé l'exigence que certaines caractéristiques soient présentes pour un ajustement pendant le processus de sélection. Les résultats de ces deux études ne sont pas directement comparables, non pas tant en raison des différences dans les algorithmes de base, mais plutôt en raison de la mise en œuvre de différentes stratégies de support.
La brève comparaison des résultats entre les deux méthodes a montré peu de chevauchement dans les pistes sur lesquelles se concentrer. On pourrait dire que le caractère commun du Ritonavir identifié par les deux méthodes fournit des preuves pour étayer la sélection de ce candidat pour une étude plus approfondie. Des études antérieures ont montré que l'utilisation d'un consensus de fonctions de notation d'amarrage protéine-ligand améliore l'identification des candidats-médicaments putatifs. 6 Pour cette raison, les chercheurs pourraient utiliser les composés uniques les mieux notés de chaque méthode pour hiérarchiser les composés pour les tests expérimentaux.
En conclusion, le criblage virtuel dérivé du pharmacophore fournit une méthode supplémentaire et complémentaire pour l'amarrage, contribuant à un consensus et à une plus grande confiance dans la sélection des candidats pour la validation expérimentale. Au-delà de l'urgence de fournir un candidat médicament à réutiliser, cette méthode basée sur les pharmacophores peut également identifier des ligands plus diversifiés pour une optimisation ultérieure des pistes.
Produits biologiques
- Patch à la nicotine
- Inventaire virtuel et impression 3D :le besoin de sécurité
- C# pour la boucle
- C pour Boucle
- Nanofibres et filaments pour une administration améliorée des médicaments
- Conception de thérapie générative
- Universal Robots lance la « plus grande » conférence virtuelle au monde sur les robots collaboratifs
- Fabrication de PCB pour la 5G
- Les micro-robots imprimés en 3D sont prometteurs pour l'administration de médicaments